Association of Host Genome with Intestinal Microbial Composition in a Large Healthy Cohort.
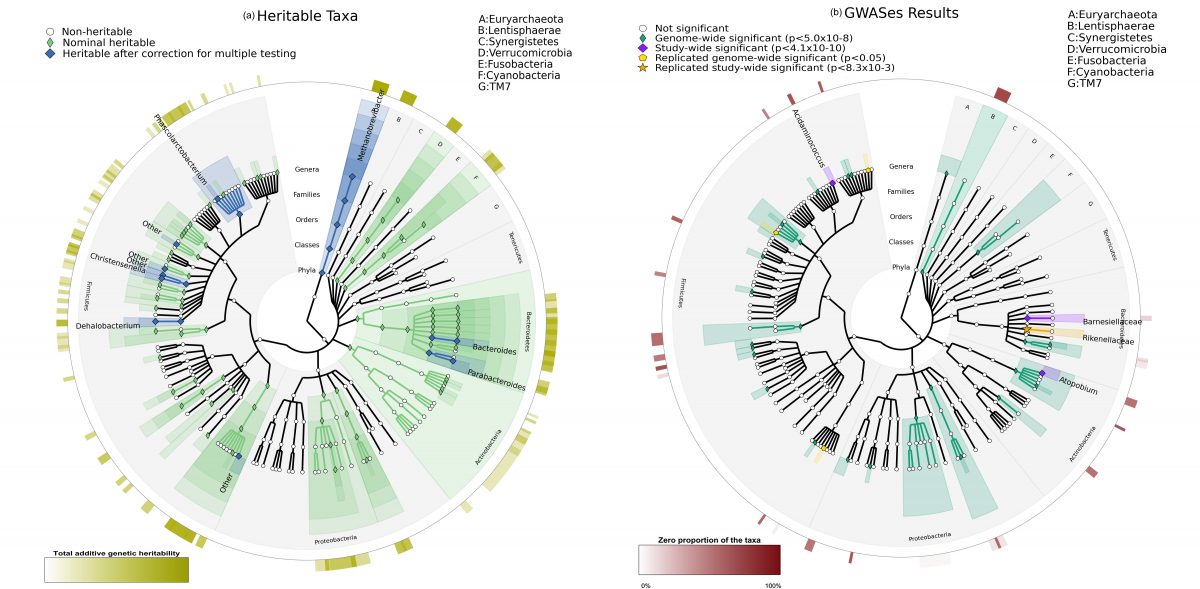
The intestinal bacteria (microbiota) are known to be influenced by the outside world, our environments, and our own bodies; the hosts. This study looks at the relationship between the intestinal microbiota (representing all bacteria living in our intestines), and the relationship these may have with our genes. Intestinal microbiota are known to be important in health and disease. Our study found that one third of the bacteria in the microbiota is heritable. This means that these heritable bacteria are more likely to be present in two individuals that are related. Our study found that 58 specific mutations in the subjects' DNA were contributing to this heritability. Their influence was observed in 33 types of the intestinal bacteria. Four of these discoveries have been confirmed in another group of subjects. This study was able to show that there are associations between specific genes and the bacteria comprising the microbiome.
Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, Guttman DS, Griffiths A, Panancionne R, Otley A, Xu L, Shestopaloff K, Moreno-Hagelsieb G, The GEM Project Research Consortium, Paterson AD, Croitoru K.
Nat Genet. 2016 Nov;48(11):1413-1417. doi: 10.1038/ng.3693. Epub 2016 Oct 3.
https://www.ncbi.nlm.nih.gov/pubmed/27694960